-
-
-
Jakarta, Indonesia
Understanding Stunted Children: Chronic Growth Failure, Early-Life Risks, and Why It Matters Beyond Height
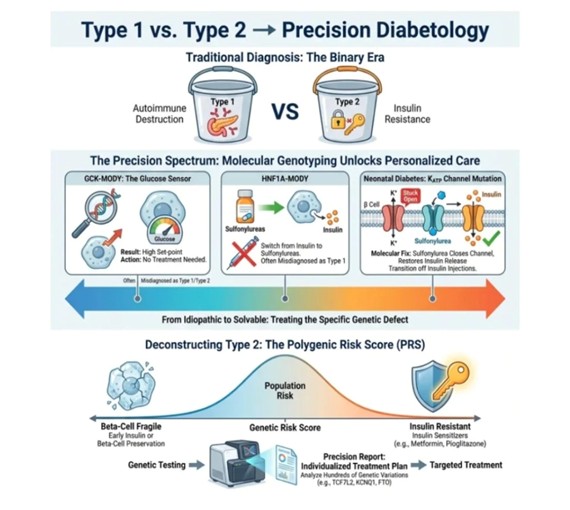
One Diagnosis, Many Diseases: The Case for Precision Diabetology
-
 Post By
Post By -
Published
March 14, 2026
The Diagnosis That Has Always Been a Shorthand
For most of modern medical history, the classification of diabetes mellitus has rested on a deceptively tidy binary. A patient either had Type 1 — the autoimmune destruction of pancreatic beta cells, demanding lifelong insulin replacement — or Type 2, characterized by progressive insulin resistance and relative secretory insufficiency, managed initially with lifestyle modification and escalating pharmacotherapy. This two-bucket model served its era well enough. It was teachable, memorable, and provided a workable framework at a time when the molecular underpinnings of glucose dysregulation were largely invisible to clinical practice.
The problem is that it was always a simplification — and not a harmless one. Patients misclassified under the Type 1 or Type 2 umbrella receive treatments calibrated for the wrong disease. They endure years, sometimes decades, on insulin regimens or oral agents that address a pathophysiology they do not actually possess, while the specific molecular defect at the root of their condition goes unidentified, untargeted, and untreated. In some cases, the correct therapy — a single oral tablet taken once daily — exists and is being withheld not out of ignorance but out of insufficient diagnostic precision.
Precision diabetology proposes a different framework. Rather than assigning patients to one of two categories based on clinical phenotype, it interrogates the molecular architecture of their glucose dysregulation: Which gene is mutated? Which pathway is disrupted? Which cellular mechanism has failed, and how specifically? The answers to these questions, now increasingly accessible through genetic sequencing and polygenic risk analysis, do not merely refine the diagnosis. They define the treatment — with a specificity and efficacy that categorical labels can never approach.
The problem is that it was always a simplification — and not a harmless one. Patients misclassified under the Type 1 or Type 2 umbrella receive treatments calibrated for the wrong disease. They endure years, sometimes decades, on insulin regimens or oral agents that address a pathophysiology they do not actually possess, while the specific molecular defect at the root of their condition goes unidentified, untargeted, and untreated. In some cases, the correct therapy — a single oral tablet taken once daily — exists and is being withheld not out of ignorance but out of insufficient diagnostic precision.
Precision diabetology proposes a different framework. Rather than assigning patients to one of two categories based on clinical phenotype, it interrogates the molecular architecture of their glucose dysregulation: Which gene is mutated? Which pathway is disrupted? Which cellular mechanism has failed, and how specifically? The answers to these questions, now increasingly accessible through genetic sequencing and polygenic risk analysis, do not merely refine the diagnosis. They define the treatment — with a specificity and efficacy that categorical labels can never approach.
The Binary Era: A Framework Built for Its Time
The Type 1 versus Type 2 distinction emerged from decades of clinical observation and immunological investigation. The autoimmune pathogenesis of Type 1 diabetes was clarified through the identification of islet-cell antibodies, the recognition of HLA-associated genetic susceptibility, and the demonstration that insulinopenia — not insulin resistance — was the primary physiological defect. Type 2 diabetes, by contrast, was framed as a disease of metabolic excess: a consequence of adiposity, sedentary behavior, and the progressive exhaustion of beta-cell reserve in the face of sustained peripheral insulin resistance.
These characterizations are not wrong, but they are incomplete in ways that matter clinically. Within the population of patients labeled as Type 2, there exist individuals whose predominant defect is beta-cell fragility rather than insulin resistance — individuals for whom metformin and lifestyle counseling offer limited benefit, and for whom early insulin initiation or beta-cell preservation strategies would be transformative. Within the population labeled as Type 1, there exist patients whose apparent autoimmune presentation conceals a monogenic defect that is both more precisely targetable and, in many cases, completely responsive to an entirely different class of drug.
The binary model, in other words, is not merely an oversimplification of biology. It is a misallocation of treatment — on a population scale, and with real consequences for patients whose molecular diagnosis has been obscured by the diagnostic categories available to their physicians.
These characterizations are not wrong, but they are incomplete in ways that matter clinically. Within the population of patients labeled as Type 2, there exist individuals whose predominant defect is beta-cell fragility rather than insulin resistance — individuals for whom metformin and lifestyle counseling offer limited benefit, and for whom early insulin initiation or beta-cell preservation strategies would be transformative. Within the population labeled as Type 1, there exist patients whose apparent autoimmune presentation conceals a monogenic defect that is both more precisely targetable and, in many cases, completely responsive to an entirely different class of drug.
The binary model, in other words, is not merely an oversimplification of biology. It is a misallocation of treatment — on a population scale, and with real consequences for patients whose molecular diagnosis has been obscured by the diagnostic categories available to their physicians.
The Precision Spectrum: When a Single Gene Explains Everything
Monogenic diabetes — diabetes caused by a mutation in a single gene — represents the clearest demonstration of what precision diabetology can achieve. These conditions, collectively estimated to account for one to four percent of all diabetes diagnoses, are systematically misdiagnosed as either Type 1 or Type 2, depending on the clinical context in which they present. The consequences of misdiagnosis are not trivial. They translate into unnecessary insulin dependence, suboptimal glycemic control, missed therapeutic opportunities, and, in some cases, a completely avoidable burden of chronic disease management.
GCK-MODY: When the Diagnosis Is the Treatment
Among the most instructive examples is glucokinase MODY, or GCK-MODY, a condition caused by a heterozygous mutation in the glucokinase gene — the enzyme that functions as the pancreatic beta cell's glucose sensor. In healthy individuals, glucokinase phosphorylates glucose at a concentration threshold of approximately five millimoles per liter, triggering insulin secretion above that set-point. In GCK-MODY, the mutation raises this threshold, so the beta cell perceives its own environment as perpetually glucose-deficient and maintains a higher-than-normal fasting glucose level as its new physiological equilibrium.
The result is a mild, stable, lifelong elevation in fasting blood glucose that is almost universally misinterpreted as early-onset Type 2 diabetes or impaired fasting glucose requiring pharmacological intervention. In reality, it requires nothing of the sort. The elevated glucose in GCK-MODY is not a sign of metabolic deterioration — it is the set-point at which the body was genetically designed to operate. Long-term cohort studies confirm that GCK-MODY carriers do not develop the microvascular complications associated with sustained hyperglycemia, and that pharmacological treatment — metformin, sulfonylureas, insulin — fails to normalize glucose because it is fighting against a molecular thermostat that is simply calibrated differently.
The clinical conclusion is counterintuitive but evidence-based: no treatment is needed. The molecular diagnosis, in this case, is the intervention. It spares the patient from unnecessary medication, unnecessary monitoring targets, and the psychological burden of being told they have a progressive metabolic disease — when, in fact, they have a benign variant in a glucose-sensing enzyme.
The result is a mild, stable, lifelong elevation in fasting blood glucose that is almost universally misinterpreted as early-onset Type 2 diabetes or impaired fasting glucose requiring pharmacological intervention. In reality, it requires nothing of the sort. The elevated glucose in GCK-MODY is not a sign of metabolic deterioration — it is the set-point at which the body was genetically designed to operate. Long-term cohort studies confirm that GCK-MODY carriers do not develop the microvascular complications associated with sustained hyperglycemia, and that pharmacological treatment — metformin, sulfonylureas, insulin — fails to normalize glucose because it is fighting against a molecular thermostat that is simply calibrated differently.
The clinical conclusion is counterintuitive but evidence-based: no treatment is needed. The molecular diagnosis, in this case, is the intervention. It spares the patient from unnecessary medication, unnecessary monitoring targets, and the psychological burden of being told they have a progressive metabolic disease — when, in fact, they have a benign variant in a glucose-sensing enzyme.
HNF1A-MODY: Switching the Lock, Not Replacing the Key
A different but equally compelling story unfolds in HNF1A-MODY, caused by mutations in the hepatocyte nuclear factor 1-alpha gene, a transcription factor critical for beta-cell development and the regulation of insulin secretory machinery. Patients with HNF1A-MODY typically present in adolescence or early adulthood with progressive hyperglycemia that, in the absence of genetic testing, is almost always attributed to Type 1 diabetes — particularly given the lean phenotype and young age of onset that characterizes many affected individuals.
The molecular defect in HNF1A-MODY impairs beta-cell insulin secretion through mechanisms that include downregulation of the KATP channel — the same potassium channel whose pharmacological closure by sulfonylureas constitutes the primary mechanism of action of that drug class. This biological coincidence has a remarkable clinical implication: patients with HNF1A-MODY are exquisitely sensitive to sulfonylureas, responding to doses far lower than those used in standard Type 2 diabetes management, and achieving glycemic control that is qualitatively superior to what insulin injections have offered them. The transition from daily or twice-daily insulin injections to a low-dose oral sulfonylurea is not merely a convenience — it is a molecular alignment of drug with defect that insulin replacement, however well titrated, cannot replicate.
The molecular defect in HNF1A-MODY impairs beta-cell insulin secretion through mechanisms that include downregulation of the KATP channel — the same potassium channel whose pharmacological closure by sulfonylureas constitutes the primary mechanism of action of that drug class. This biological coincidence has a remarkable clinical implication: patients with HNF1A-MODY are exquisitely sensitive to sulfonylureas, responding to doses far lower than those used in standard Type 2 diabetes management, and achieving glycemic control that is qualitatively superior to what insulin injections have offered them. The transition from daily or twice-daily insulin injections to a low-dose oral sulfonylurea is not merely a convenience — it is a molecular alignment of drug with defect that insulin replacement, however well titrated, cannot replicate.
Neonatal Diabetes: Closing a Channel, Opening a New Life
Perhaps the most dramatic illustration of precision diabetology's potential lies in neonatal diabetes caused by activating mutations in the KCNJ11 or ABCC8 genes, which encode the two subunits of the KATP channel itself. In normal beta-cell physiology, rising intracellular ATP — a consequence of glucose metabolism — closes the KATP channel, depolarizes the cell membrane, opens voltage-gated calcium channels, and triggers insulin exocytosis. In neonatal diabetes caused by these gain-of-function mutations, the KATP channel is locked open, resistant to the ATP signal, and perpetually blocking the insulin release cascade. The beta cells are structurally intact. They sense glucose. They metabolize it normally. They simply cannot complete the final signaling step that would release insulin into the circulation.
For decades, these infants were diagnosed with Type 1 diabetes and placed on insulin therapy — not because insulin was the right treatment, but because it was the only framework available. The discovery that sulfonylureas could close the mutant KATP channel by binding to the SUR1 subunit at a site independent of the ATP-binding domain represented one of the most elegant therapeutic translations in the history of endocrinology. Clinical trials demonstrated that the majority of patients with KCNJ11 or ABCC8 neonatal diabetes could be successfully transitioned from insulin injections to oral sulfonylurea therapy — in many cases achieving better glycemic control, with lower HbA1c values and fewer hypoglycemic episodes, than they had ever achieved on insulin.
The word 'transition' understates what this represents for the patients and families involved. For a child diagnosed with apparent Type 1 diabetes in infancy, the prospect of growing up free from insulin injections — free from pump tubing, continuous glucose monitors, and the constant arithmetic of carbohydrate counting — is not a therapeutic incremental improvement. It is a transformation in the lived experience of disease. It is what becomes possible when the diagnosis is made at the molecular level and the treatment is matched to the mechanism.
For decades, these infants were diagnosed with Type 1 diabetes and placed on insulin therapy — not because insulin was the right treatment, but because it was the only framework available. The discovery that sulfonylureas could close the mutant KATP channel by binding to the SUR1 subunit at a site independent of the ATP-binding domain represented one of the most elegant therapeutic translations in the history of endocrinology. Clinical trials demonstrated that the majority of patients with KCNJ11 or ABCC8 neonatal diabetes could be successfully transitioned from insulin injections to oral sulfonylurea therapy — in many cases achieving better glycemic control, with lower HbA1c values and fewer hypoglycemic episodes, than they had ever achieved on insulin.
The word 'transition' understates what this represents for the patients and families involved. For a child diagnosed with apparent Type 1 diabetes in infancy, the prospect of growing up free from insulin injections — free from pump tubing, continuous glucose monitors, and the constant arithmetic of carbohydrate counting — is not a therapeutic incremental improvement. It is a transformation in the lived experience of disease. It is what becomes possible when the diagnosis is made at the molecular level and the treatment is matched to the mechanism.
Deconstructing Type 2: The Polygenic Risk Score Revolution
Monogenic diabetes, while paradigmatically important, accounts for a small minority of all diabetes diagnoses. The far larger challenge lies in deconstructing Type 2 diabetes itself — a heterogeneous condition that, under a single diagnostic label, encompasses a spectrum of genetic architectures, primary pathophysiological mechanisms, and therefore optimal therapeutic strategies. This is where polygenic risk scoring enters the precision diabetology framework.
Beyond 'Type 2': A Spectrum Within a Spectrum
Unlike monogenic diabetes, where a single mutant gene explains the entire phenotype, Type 2 diabetes arises from the cumulative effect of hundreds or thousands of common genetic variants, each contributing a small increment to overall risk. Genome-wide association studies have identified dozens of loci consistently associated with Type 2 diabetes risk, including variants in TCF7L2 (which encodes a transcription factor critical for beta-cell function), KCNQ1 (which encodes a potassium channel involved in insulin secretion), and FTO (a gene associated with obesity and energy homeostasis). Individually, each of these variants exerts a modest effect. Collectively, in the form of a polygenic risk score, they describe the genetic architecture of an individual's metabolic vulnerability with a granularity that clinical phenotyping alone cannot achieve.
Crucially, not all polygenic risk scores for Type 2 diabetes cluster around the same biological mechanisms. Research using pathway-partitioned polygenic scores has revealed at least two major genetic subtypes within the Type 2 diabetes population: those whose genetic risk is concentrated in variants affecting beta-cell function and survival, and those whose genetic risk is driven primarily by variants associated with insulin resistance, adiposity, and lipid metabolism. These are not the same disease. They should not be treated as though they are.
Crucially, not all polygenic risk scores for Type 2 diabetes cluster around the same biological mechanisms. Research using pathway-partitioned polygenic scores has revealed at least two major genetic subtypes within the Type 2 diabetes population: those whose genetic risk is concentrated in variants affecting beta-cell function and survival, and those whose genetic risk is driven primarily by variants associated with insulin resistance, adiposity, and lipid metabolism. These are not the same disease. They should not be treated as though they are.
The Fragile Beta-Cell Subtype: Early Protection Over Late Replacement
Patients whose genetic risk profile is dominated by beta-cell fragility variants present a fundamentally different therapeutic imperative than the insulin-resistance-predominant subtype. Their pancreatic reserve is constitutionally limited, and the natural history of their disease is one of earlier, faster progression toward insulin dependency — a progression that standard metformin therapy, which has no direct beta-cell protective effect, does little to arrest. For these individuals, the precision diabetology approach recommends early insulin therapy — not as a last resort after oral agent failure, but as a proactive strategy to reduce the metabolic demand on a beta-cell mass that is inherently insufficient — or alternatively, the deployment of agents with established beta-cell preservation properties, such as GLP-1 receptor agonists, which have demonstrated antiapoptotic effects on beta cells in multiple preclinical and early clinical studies.
The clinical logic here is preventive: by protecting a fragile beta-cell mass before it has been exhausted, rather than replacing its secretory output after it has failed, we may be able to substantially alter the natural history of this subtype of Type 2 diabetes. This is a fundamentally different therapeutic goal than symptom management, and it requires identifying the fragile-beta-cell patient before their islet reserve has been depleted — a task for which genetic risk scoring is uniquely suited.
The clinical logic here is preventive: by protecting a fragile beta-cell mass before it has been exhausted, rather than replacing its secretory output after it has failed, we may be able to substantially alter the natural history of this subtype of Type 2 diabetes. This is a fundamentally different therapeutic goal than symptom management, and it requires identifying the fragile-beta-cell patient before their islet reserve has been depleted — a task for which genetic risk scoring is uniquely suited.
The Insulin-Resistant Subtype: Targeting the Primary Defect
At the other end of the polygenic spectrum, patients whose risk architecture is predominantly insulin-resistance-driven represent a population for whom insulin sensitizers are the logical first-line therapy. Metformin, which reduces hepatic glucose production and improves peripheral insulin sensitivity, has served this population well for decades. Thiazolidinediones such as pioglitazone, despite their fallen status in routine practice due to concerns about weight gain and fluid retention, remain among the most potent insulin sensitizers available and may represent the appropriate pharmacological choice for patients whose genotype identifies insulin resistance as the dominant pathophysiological defect.
For this subtype, the precision approach also reframes the role of lifestyle intervention. Physical activity and dietary modification improve insulin sensitivity through mechanisms that directly address the genetic risk driving this subtype's pathophysiology — a more mechanistically coherent rationale for lifestyle prescription than the generic advice to 'eat less and move more' that is currently delivered without distinction to all newly diagnosed Type 2 patients regardless of their biological subtype.
For this subtype, the precision approach also reframes the role of lifestyle intervention. Physical activity and dietary modification improve insulin sensitivity through mechanisms that directly address the genetic risk driving this subtype's pathophysiology — a more mechanistically coherent rationale for lifestyle prescription than the generic advice to 'eat less and move more' that is currently delivered without distinction to all newly diagnosed Type 2 patients regardless of their biological subtype.
The Precision Report: From Sequencing to Prescription
The practical instrument through which polygenic risk is translated into clinical decision-making is what precision diabetology envisions as the individualized precision report — a comprehensive genetic analysis that integrates polygenic risk scores across hundreds of disease-relevant variants, identifies the dominant pathophysiological subtype, and generates a treatment recommendation grounded in the patient's molecular profile rather than their phenotypic presentation alone. The genetic testing pipeline required to generate this report is increasingly accessible, cost-effective, and interpretable, particularly as reference population databases expand and computational risk modeling improves.
The precision report does not replace clinical judgment. Phenotypic factors — body weight, age of onset, family history, comorbidities, medication tolerability — remain integral to treatment decisions. What the precision report provides is a molecular layer of information that clinical assessment alone cannot supply, and that has the potential to meaningfully alter the therapeutic trajectory for a substantial proportion of patients currently receiving phenotype-based care that is not optimized for their specific biological defect.
The precision report does not replace clinical judgment. Phenotypic factors — body weight, age of onset, family history, comorbidities, medication tolerability — remain integral to treatment decisions. What the precision report provides is a molecular layer of information that clinical assessment alone cannot supply, and that has the potential to meaningfully alter the therapeutic trajectory for a substantial proportion of patients currently receiving phenotype-based care that is not optimized for their specific biological defect.
The Misdiagnosis Crisis: A Problem Precision Can Solve
Running as a thread through every layer of the precision diabetology framework is a problem that the medical community has been slow to confront at scale: the misdiagnosis of diabetes is not a rare event. It is, by any reasonable estimate, a systematic occurrence affecting tens of thousands of patients worldwide. GCK-MODY patients are told they have impaired fasting glucose and commenced on metformin. HNF1A-MODY patients presenting in their teens with rapid-onset hyperglycemia are diagnosed with Type 1 and spend years on insulin they do not need. Neonatal diabetes patients, perhaps the most poignantly misdiagnosed of all, are given the most intensive available therapy for a disease they do not have, while the specific molecular defect that would respond to a simple oral agent goes undetected.
The barriers to genetic testing in diabetes are genuine and should not be minimized. Access to molecular diagnostics varies enormously by geography, health system structure, and reimbursement policy. Clinical awareness of the monogenic diabetes subtypes remains patchy even among endocrinologists, and the referral pathways for genetic testing are frequently absent or poorly defined. These are solvable problems — but solving them requires an active commitment by the diabetes community to treat molecular diagnosis not as an academic luxury but as a clinical obligation for patients whose presentation raises even the possibility of a monogenic etiology.
Clinical phenotypic triggers — young age of onset, absence of autoantibodies, strong family history following a dominant inheritance pattern, unusual drug sensitivity, or atypical glycemic patterns — should serve as indications for genetic evaluation. Guidelines from the European and American diabetes associations increasingly reflect this, though implementation in routine practice lags meaningfully behind the evidence base.
The barriers to genetic testing in diabetes are genuine and should not be minimized. Access to molecular diagnostics varies enormously by geography, health system structure, and reimbursement policy. Clinical awareness of the monogenic diabetes subtypes remains patchy even among endocrinologists, and the referral pathways for genetic testing are frequently absent or poorly defined. These are solvable problems — but solving them requires an active commitment by the diabetes community to treat molecular diagnosis not as an academic luxury but as a clinical obligation for patients whose presentation raises even the possibility of a monogenic etiology.
Clinical phenotypic triggers — young age of onset, absence of autoantibodies, strong family history following a dominant inheritance pattern, unusual drug sensitivity, or atypical glycemic patterns — should serve as indications for genetic evaluation. Guidelines from the European and American diabetes associations increasingly reflect this, though implementation in routine practice lags meaningfully behind the evidence base.
Precision Diabetology in Practice: Where Are We Now?
The transition from binary classification to precision diabetology is not a future aspiration — it is an ongoing clinical reality, though unevenly distributed. Specialist centers in the United Kingdom, the United States, Denmark, and Australia have developed monogenic diabetes diagnostic services that now function as referral hubs for patients whose clinical features suggest a single-gene etiology. The MODY probability calculator, developed from population data, allows clinicians to estimate the pre-test probability of monogenic diabetes based on clinical parameters and guide appropriate referral for sequencing.
Polygenic risk scoring for Type 2 diabetes stratification remains primarily a research tool at present, though its translation into clinical practice is accelerating. Several commercial genetic testing platforms already incorporate diabetes-relevant polygenic scores into their reports, and the integration of genomic data into electronic health records — already underway at a small number of health systems — creates the infrastructure for population-level precision prescribing that is the ultimate expression of the framework described here.
The challenges that remain are primarily those of implementation: training clinicians to recognize phenotypic triggers for genetic testing, establishing reimbursement frameworks that make sequencing accessible outside academic centers, developing computational tools that translate polygenic scores into clinically actionable treatment recommendations, and building the health system infrastructure to deliver precision diabetology not as a specialist privilege but as a routine standard of care.
Polygenic risk scoring for Type 2 diabetes stratification remains primarily a research tool at present, though its translation into clinical practice is accelerating. Several commercial genetic testing platforms already incorporate diabetes-relevant polygenic scores into their reports, and the integration of genomic data into electronic health records — already underway at a small number of health systems — creates the infrastructure for population-level precision prescribing that is the ultimate expression of the framework described here.
The challenges that remain are primarily those of implementation: training clinicians to recognize phenotypic triggers for genetic testing, establishing reimbursement frameworks that make sequencing accessible outside academic centers, developing computational tools that translate polygenic scores into clinically actionable treatment recommendations, and building the health system infrastructure to deliver precision diabetology not as a specialist privilege but as a routine standard of care.
Conclusion: From Idiopathic to Solvable
The figure that opens this article carries a subtitle that deserves to be read carefully: from idiopathic to solvable. For decades, the heterogeneity of the diabetes phenotype was acknowledged but largely accommodated within a framework that lacked the diagnostic tools to resolve it. Patients whose disease did not behave as expected were managed with escalating empiricism — more drugs, higher doses, closer monitoring — without a clear understanding of why the standard approach was failing them.
Precision diabetology offers a different answer to that question. It says that the failure is not in the patient, and not in the clinician, but in the diagnostic framework — one that was built before the molecular tools existed to interrogate it more deeply. The bucket of Type 1 and the bucket of Type 2 were always containers for clinical patterns, not for biological mechanisms. Now that we have the tools to see the mechanisms directly, the buckets become insufficient.
What replaces them is not complexity for its own sake — it is clarity. The GCK-MODY patient who needs no medication. The neonatal diabetes infant who can live without insulin. The high-risk beta-cell-fragile patient who benefits from early protection rather than late rescue. The insulin-resistant patient in whom metformin targets precisely the right mechanism. These are not niche academic distinctions. They are clinical realities waiting to be identified — and in an era of accessible genomic sequencing, there is diminishing justification for allowing the diagnostic binary to obscure them.
Precision diabetology is not the end of the Type 1 and Type 2 framework. It is its maturation — an acknowledgment that the categories our predecessors created served their purpose, and that the purpose now demands greater resolution. The molecular era of diabetes medicine has arrived. The patients who will benefit from it most are those who never quite fit the buckets to begin with.
Precision diabetology offers a different answer to that question. It says that the failure is not in the patient, and not in the clinician, but in the diagnostic framework — one that was built before the molecular tools existed to interrogate it more deeply. The bucket of Type 1 and the bucket of Type 2 were always containers for clinical patterns, not for biological mechanisms. Now that we have the tools to see the mechanisms directly, the buckets become insufficient.
What replaces them is not complexity for its own sake — it is clarity. The GCK-MODY patient who needs no medication. The neonatal diabetes infant who can live without insulin. The high-risk beta-cell-fragile patient who benefits from early protection rather than late rescue. The insulin-resistant patient in whom metformin targets precisely the right mechanism. These are not niche academic distinctions. They are clinical realities waiting to be identified — and in an era of accessible genomic sequencing, there is diminishing justification for allowing the diagnostic binary to obscure them.
Precision diabetology is not the end of the Type 1 and Type 2 framework. It is its maturation — an acknowledgment that the categories our predecessors created served their purpose, and that the purpose now demands greater resolution. The molecular era of diabetes medicine has arrived. The patients who will benefit from it most are those who never quite fit the buckets to begin with.
Urinary Tract Infection Updates: Current Clinical and Public Health Developments
Urinary tract infection remains one of the most common bacterial infections globally, with current updates focusing on antimicrobial resistance, diagnostic stewardship, recurrent infection management, catheter-associated prevention, and emerging non-antibiotic strategies. This article reviews recent developments relevant to clinicians, researchers, and public health professionals.
Top 5 Vegetables That May Enhance Immune Function: An Evidence-Based Nutritional Overview
Dietary patterns rich in vegetables are consistently associated with improved health outcomes, including support of normal immune function. This article reviews five vegetables—broccoli, spinach, garlic, carrots, and red bell peppers—that may contribute to immune resilience through their content of vitamins, minerals, antioxidants, and bioactive phytochemicals.


Understanding Stunted Children: Chronic Growth Failure, Early-Life Risks, and Why It Matters Beyond Height
Stunting in children refers to impaired linear growth resulting from chronic undernutrition, repeated infection, and unfavorable early-life conditions. More than a matter of short stature, stunting reflects a broader process of biological and developmental disadvantage that can affect cognitive outcomes, school performance, and long-term health.
Blood Pressure Monitoring as a Public Health Priority: Strengthening Early Detection and Long-Term Cardiovascular Prevention
Blood pressure monitoring remains one of the most practical and impactful tools in public health for identifying hypertension early, guiding treatment decisions, and reducing long-term cardiovascular risk. Wider adoption of accurate office, community, and home-based monitoring strategies could significantly improve prevention of stroke, heart disease, kidney damage, and premature mortality.

