-
-
-
Jakarta, Indonesia
Understanding Stunted Children: Chronic Growth Failure, Early-Life Risks, and Why It Matters Beyond Height
The Silent Atrophy: Why We Are Rewriting Rules on Major Depressive Disorders?
-
 Post By
Post By -
Published
March 14, 2026
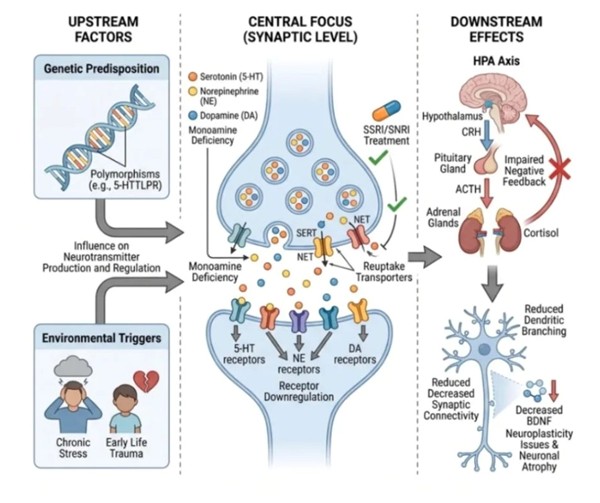
Upstream Factors: The Genetic and Environmental Seeds of Depression
Major Depressive Disorder does not arise from a single cause, and one of the most important corrections that contemporary psychiatry has made to the public's understanding of the illness is the rejection of any monocausal explanation. Depression is not simply the result of a difficult life event, nor is it purely a matter of inherited biology. Instead, the evidence from decades of genetic epidemiology, neuroimaging, and longitudinal developmental research converges on a diathesis-stress model: the condition emerges from the interaction between an individual's biological vulnerability and the specific stressors their environment imposes. On the genetic side, twin studies consistently demonstrate heritability estimates of approximately 37% for major depression, indicating that a substantial but non-deterministic portion of risk is encoded in the genome. Among the most extensively studied genetic variants is the 5-HTTLPR polymorphism — a length variation in the promoter region of the SLC6A4 gene encoding the serotonin transporter — which modulates the efficiency of serotonin reuptake and has been associated with heightened emotional reactivity and increased susceptibility to depression in the context of adverse life events. This gene-environment interaction is the key: the genetic variant does not cause depression in isolation, but it does calibrate the nervous system's sensitivity in ways that matter when environmental adversity arrives. That adversity most commonly takes the form of early life trauma — childhood abuse, neglect, or loss — and chronic adult psychosocial stress, both of which program lasting dysregulation of the neuroendocrine and neurotransmitter systems that these same genetic variants influence. The upstream factors do not cause depression directly; they configure the biological substrate upon which downstream pathology unfolds.
The Synaptic Level: Monoamine Deficiency and the Pharmacology of Hope
For much of the latter half of the twentieth century, the monoamine hypothesis stood as the dominant explanatory framework for depression — and while it has since been substantially refined, it remains the most clinically actionable description of what happens at the neural level in the acutely depressed brain. The hypothesis, in its original form, proposed that depression results from a functional deficiency of monoamine neurotransmitters — principally serotonin (5-HT), norepinephrine (NE), and dopamine (DA) — in the synaptic cleft: the microscopic gap between neurons across which chemical signals must travel for intercellular communication to occur. When concentrations of these monoamines are insufficient, the signal fails to cross effectively, and the complex affective, motivational, and cognitive processes that depend on intact monoaminergic neurotransmission — mood regulation, hedonic capacity, reward anticipation, energy, concentration, and psychomotor function — begin to deteriorate. A second, compounding mechanism is receptor downregulation: chronic exposure to low neurotransmitter concentrations causes the post-synaptic neuron to reduce the density or sensitivity of its receptors as a compensatory adaptation, further impairing the signal even in response to whatever monoamine is present. It is precisely at this synaptic level that the most widely prescribed antidepressants intervene. Selective serotonin reuptake inhibitors (SSRIs) and serotonin-norepinephrine reuptake inhibitors (SNRIs) block the reuptake transporters — SERT for serotonin and NET for norepinephrine — that would ordinarily clear monoamines from the synapse after they are released. By occupying these transporters, SSRIs and SNRIs ensure that whatever serotonin or norepinephrine is released lingers in the synaptic cleft longer than it otherwise would, increasing the probability that it will bind a post-synaptic receptor and successfully propagate the signal. This pharmacological mechanism is not a cure; it does not address the upstream factors or the downstream structural changes. But for the majority of patients, it represents a meaningful and evidence-based modulation of the most accessible and functionally critical point in the depressive cascade.
Downstream Effects: The Silent Atrophy of the Untreated Depressed Brain
The most sobering insight from two decades of neuroimaging and neuroendocrinological research in depression is that the disease is not merely a functional disorder — it is, in its chronic and undertreated forms, a condition that inflicts measurable structural damage on the brain. The primary mechanism of this structural harm is cortisol neurotoxicity, mediated through dysregulation of the hypothalamic-pituitary-adrenal axis. Under normal physiological conditions, the HPA axis operates through a tightly regulated negative feedback loop: the hypothalamus releases corticotropin-releasing hormone (CRH), which stimulates the pituitary to release adrenocorticotropic hormone (ACTH), which in turn drives the adrenal glands to secrete cortisol. Cortisol then feeds back to both the hypothalamus and pituitary, suppressing further CRH and ACTH release, and maintaining circadian cortisol rhythms within a narrow healthy range. In individuals with depression — particularly those whose upstream vulnerability includes a history of early trauma — this negative feedback loop is impaired. The hippocampus, which is richly endowed with glucocorticoid receptors and normally participates in HPA suppression, loses its inhibitory influence as chronic hypercortisolemia progressively damages its glucocorticoid receptor population. The result is a self-perpetuating cycle of cortisol excess that the system can no longer self-correct. Sustained elevation of cortisol exerts direct neurotoxic effects on hippocampal neurons through multiple mechanisms: it suppresses the expression of brain-derived neurotrophic factor (BDNF), a protein essential for neuronal survival, dendritic growth, and synaptic plasticity; it promotes glutamate excitotoxicity through NMDA receptor overactivation; and it triggers apoptotic cascades in vulnerable neuronal populations. The macroscopic consequence of these cellular insults is measurable: volumetric MRI studies have consistently demonstrated hippocampal volume reduction in patients with recurrent or chronic major depression, with greater volume loss correlating with longer illness duration and greater number of depressive episodes. This finding has fundamentally shifted the clinical imperative around depression treatment — it is no longer sufficient to view each depressive episode as a transient functional state that resolves between episodes. Each episode represents a period during which neurotoxic processes are operating, dendritic branching is being reduced, synaptic connectivity is declining, and the structural substrate of cognition and emotional regulation is being quietly eroded. Early, sustained, and effective treatment — addressing both the synaptic level and the upstream drivers — is therefore not simply about relieving suffering in the present. It is about protecting the architecture of the brain for the future.
Urinary Tract Infection Updates: Current Clinical and Public Health Developments
Urinary tract infection remains one of the most common bacterial infections globally, with current updates focusing on antimicrobial resistance, diagnostic stewardship, recurrent infection management, catheter-associated prevention, and emerging non-antibiotic strategies. This article reviews recent developments relevant to clinicians, researchers, and public health professionals.
Top 5 Vegetables That May Enhance Immune Function: An Evidence-Based Nutritional Overview
Dietary patterns rich in vegetables are consistently associated with improved health outcomes, including support of normal immune function. This article reviews five vegetables—broccoli, spinach, garlic, carrots, and red bell peppers—that may contribute to immune resilience through their content of vitamins, minerals, antioxidants, and bioactive phytochemicals.


Understanding Stunted Children: Chronic Growth Failure, Early-Life Risks, and Why It Matters Beyond Height
Stunting in children refers to impaired linear growth resulting from chronic undernutrition, repeated infection, and unfavorable early-life conditions. More than a matter of short stature, stunting reflects a broader process of biological and developmental disadvantage that can affect cognitive outcomes, school performance, and long-term health.
Blood Pressure Monitoring as a Public Health Priority: Strengthening Early Detection and Long-Term Cardiovascular Prevention
Blood pressure monitoring remains one of the most practical and impactful tools in public health for identifying hypertension early, guiding treatment decisions, and reducing long-term cardiovascular risk. Wider adoption of accurate office, community, and home-based monitoring strategies could significantly improve prevention of stroke, heart disease, kidney damage, and premature mortality.

