-
-
-
Jakarta, Indonesia
Understanding Stunted Children: Chronic Growth Failure, Early-Life Risks, and Why It Matters Beyond Height
The Great Imitator: Decoding the Chaos of Systemic Lupus Erythematosus (SLE)
-
 Post By
Post By -
Published
March 8, 2026
The Great Imitator at Its Most Personal
Among the autoimmune diseases, systemic lupus erythematosus has long carried the epithet of the great imitator — a condition whose mercurial clinical manifestations can mimic virtually any inflammatory or infectious disease in medicine, frustrating diagnosis, delaying treatment, and accumulating organ damage in the interval between first symptom and correct identification. Nowhere is this mimicry more immediately visible, and paradoxically more frequently misread, than in the skin. The cutaneous manifestations of lupus are not merely cosmetic accompaniments to a systemic disease. They are, in many patients, its first and most prominent expression — preceding the development of systemic lupus erythematosus by months or years, occurring in isolation as purely cutaneous disease, or serving as real-time indicators of systemic disease activity.
Cutaneous lupus erythematosus encompasses a heterogeneous group of autoimmune skin disorders united by a common pathogenetic mechanism but distinguished by their clinical morphology, anatomical distribution, depth of tissue involvement, propensity for scarring, and relationship to systemic disease. Understanding these subtypes — and, more fundamentally, understanding the molecular events that generate them — is essential not only for accurate diagnosis but for rational therapeutic decision-making in an era where targeted immunological interventions are increasingly available.
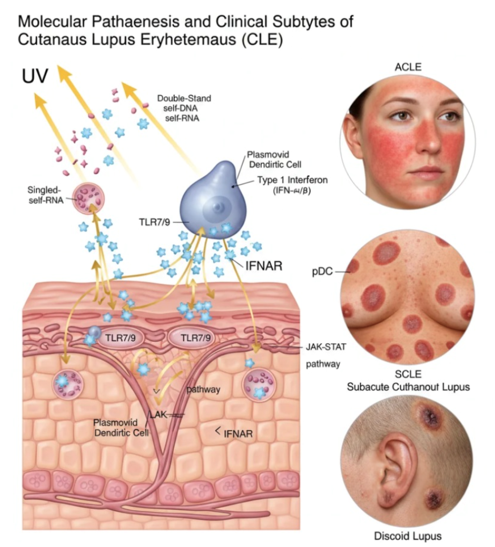
The figure that opens this article presents the pathogenetic cascade and its clinical consequences within a single integrated framework. This article traces that cascade from its initiating environmental trigger — ultraviolet light — through the molecular machinery of innate immune activation, interferon amplification, and intracellular signaling, to the three principal clinical subtypes that emerge from this shared biological substrate: acute cutaneous lupus erythematosus, subacute cutaneous lupus erythematosus, and discoid lupus erythematosus.
Cutaneous lupus erythematosus encompasses a heterogeneous group of autoimmune skin disorders united by a common pathogenetic mechanism but distinguished by their clinical morphology, anatomical distribution, depth of tissue involvement, propensity for scarring, and relationship to systemic disease. Understanding these subtypes — and, more fundamentally, understanding the molecular events that generate them — is essential not only for accurate diagnosis but for rational therapeutic decision-making in an era where targeted immunological interventions are increasingly available.
The figure that opens this article presents the pathogenetic cascade and its clinical consequences within a single integrated framework. This article traces that cascade from its initiating environmental trigger — ultraviolet light — through the molecular machinery of innate immune activation, interferon amplification, and intracellular signaling, to the three principal clinical subtypes that emerge from this shared biological substrate: acute cutaneous lupus erythematosus, subacute cutaneous lupus erythematosus, and discoid lupus erythematosus.
The Environmental Trigger: Ultraviolet Light and the Release of Self
The relationship between ultraviolet radiation and lupus has been recognized clinically for more than a century — patients with lupus are photosensitive, their disease flares with sun exposure, and their skin lesions preferentially involve sun-exposed anatomical sites. What was long observed empirically is now understood mechanistically, and the mechanism is both elegant and deeply counterintuitive: UV light triggers lupus not by introducing a foreign pathogen, but by causing the body's own cells to release fragments of themselves that the immune system then mistakes for an invader.
When ultraviolet light — particularly UVB, the shorter-wavelength component of the solar spectrum that is most efficiently absorbed by DNA — strikes the keratinocytes of the epidermis, it inflicts direct photochemical damage on the cellular genome. DNA strand breaks, pyrimidine dimer formation, and the oxidative modification of RNA molecules are among the immediate molecular consequences of this photodamage. Under normal circumstances, these injured cells would undergo regulated apoptosis — a tidy, immunologically silent form of cell death in which the cellular contents are packaged into membrane-enclosed apoptotic bodies and cleared by phagocytes without triggering inflammation.
In lupus, this clearance mechanism is defective. Apoptotic bodies accumulate in UV-damaged skin rather than being efficiently phagocytosed, and their contents — including double-stranded DNA, single-stranded RNA, ribonucleoproteins, and other nuclear material — leak into the extracellular space of the dermis and epidermis. These nucleic acid fragments are not, in a healthy immune system, particularly alarming: the immune system has evolved an array of tolerance mechanisms that prevent self-nucleic acids from triggering inflammation. In lupus, those tolerance mechanisms are compromised, and the liberated self-DNA and self-RNA become the molecular kindling for a self-perpetuating autoimmune fire.
The downstream consequences of impaired apoptotic clearance extend beyond the immediate UV-exposed site. Autoantibodies against nuclear antigens — anti-dsDNA, anti-Sm, anti-Ro/SSA, anti-La/SSB — which are pathognomonic features of systemic lupus, bind to the extracellular nuclear debris and form immune complexes that are themselves potent activators of the innate immune machinery. The released nucleic acids and their immune complexes are then taken up by the critical cellular intermediary in the cutaneous lupus pathogenetic cascade: the plasmacytoid dendritic cell.
When ultraviolet light — particularly UVB, the shorter-wavelength component of the solar spectrum that is most efficiently absorbed by DNA — strikes the keratinocytes of the epidermis, it inflicts direct photochemical damage on the cellular genome. DNA strand breaks, pyrimidine dimer formation, and the oxidative modification of RNA molecules are among the immediate molecular consequences of this photodamage. Under normal circumstances, these injured cells would undergo regulated apoptosis — a tidy, immunologically silent form of cell death in which the cellular contents are packaged into membrane-enclosed apoptotic bodies and cleared by phagocytes without triggering inflammation.
In lupus, this clearance mechanism is defective. Apoptotic bodies accumulate in UV-damaged skin rather than being efficiently phagocytosed, and their contents — including double-stranded DNA, single-stranded RNA, ribonucleoproteins, and other nuclear material — leak into the extracellular space of the dermis and epidermis. These nucleic acid fragments are not, in a healthy immune system, particularly alarming: the immune system has evolved an array of tolerance mechanisms that prevent self-nucleic acids from triggering inflammation. In lupus, those tolerance mechanisms are compromised, and the liberated self-DNA and self-RNA become the molecular kindling for a self-perpetuating autoimmune fire.
The downstream consequences of impaired apoptotic clearance extend beyond the immediate UV-exposed site. Autoantibodies against nuclear antigens — anti-dsDNA, anti-Sm, anti-Ro/SSA, anti-La/SSB — which are pathognomonic features of systemic lupus, bind to the extracellular nuclear debris and form immune complexes that are themselves potent activators of the innate immune machinery. The released nucleic acids and their immune complexes are then taken up by the critical cellular intermediary in the cutaneous lupus pathogenetic cascade: the plasmacytoid dendritic cell.
The Plasmacytoid Dendritic Cell: Mistaking Self for Non-Self
The plasmacytoid dendritic cell is a specialized innate immune cell whose primary evolutionary purpose is the detection of viral infection and the rapid, massive production of Type I interferons in response to viral nucleic acids. This specialization is mediated through the expression of Toll-like receptors 7 and 9 — endosomal pattern recognition receptors that are activated by single-stranded RNA (TLR7) and unmethylated CpG-containing DNA (TLR9), molecular signatures that are characteristic of viral genomes.
The immunological tragedy of cutaneous lupus is that the self-nucleic acids released by UV-damaged keratinocytes — particularly when complexed with autoantibodies that facilitate their uptake into endosomal compartments via Fc receptor-mediated internalization — bear sufficient molecular resemblance to viral nucleic acids to trigger TLR7 and TLR9 activation within the plasmacytoid dendritic cell. The cellular machinery cannot discriminate between the DNA of an invading pathogen and the double-stranded DNA of the patient's own damaged cells. The receptor binds its ligand. The signaling cascade is initiated. The plasmacytoid dendritic cell responds as though it has detected a viral emergency.
The result is the production of Type I interferons — specifically interferon-alpha and interferon-beta — in quantities that are vastly disproportionate to any genuine infectious threat. Plasmacytoid dendritic cells are among the most prolific Type I interferon producers in the entire immune system, capable of secreting thousands of times more interferon per cell than other innate immune populations. In the UV-exposed skin of a lupus patient, this response is both amplified by the ongoing nucleic acid stimulus and unconstrained by the negative feedback mechanisms that would normally terminate it, creating a self-sustaining loop of innate immune activation.
The presence of plasmacytoid dendritic cells in the lesional skin of all major cutaneous lupus subtypes — and their relative absence in the non-lesional skin of the same patients — has been confirmed by immunohistochemistry and transcriptional profiling studies, establishing these cells as not merely incidental bystanders but active pathological participants in the cutaneous inflammatory process. Their tissue localization — particularly along the dermal-epidermal junction and within the perivascular dermis — places them precisely at the anatomical interface where the inflammatory damage of cutaneous lupus is most characteristically expressed.
The immunological tragedy of cutaneous lupus is that the self-nucleic acids released by UV-damaged keratinocytes — particularly when complexed with autoantibodies that facilitate their uptake into endosomal compartments via Fc receptor-mediated internalization — bear sufficient molecular resemblance to viral nucleic acids to trigger TLR7 and TLR9 activation within the plasmacytoid dendritic cell. The cellular machinery cannot discriminate between the DNA of an invading pathogen and the double-stranded DNA of the patient's own damaged cells. The receptor binds its ligand. The signaling cascade is initiated. The plasmacytoid dendritic cell responds as though it has detected a viral emergency.
The result is the production of Type I interferons — specifically interferon-alpha and interferon-beta — in quantities that are vastly disproportionate to any genuine infectious threat. Plasmacytoid dendritic cells are among the most prolific Type I interferon producers in the entire immune system, capable of secreting thousands of times more interferon per cell than other innate immune populations. In the UV-exposed skin of a lupus patient, this response is both amplified by the ongoing nucleic acid stimulus and unconstrained by the negative feedback mechanisms that would normally terminate it, creating a self-sustaining loop of innate immune activation.
The presence of plasmacytoid dendritic cells in the lesional skin of all major cutaneous lupus subtypes — and their relative absence in the non-lesional skin of the same patients — has been confirmed by immunohistochemistry and transcriptional profiling studies, establishing these cells as not merely incidental bystanders but active pathological participants in the cutaneous inflammatory process. Their tissue localization — particularly along the dermal-epidermal junction and within the perivascular dermis — places them precisely at the anatomical interface where the inflammatory damage of cutaneous lupus is most characteristically expressed.
The Interferon Cascade: From Signal to Inflammation Type I Interferons and the IFNAR Receptor
The Type I interferons secreted by activated plasmacytoid dendritic cells do not act locally and transiently, as cytokines in a regulated acute inflammatory response are designed to do. They bind to the interferon-alpha/beta receptor — IFNAR — which is expressed ubiquitously on virtually all nucleated cells in the dermis and epidermis, as well as on circulating immune cells that traffic through the skin. IFNAR is a heterodimeric cell-surface receptor composed of IFNAR1 and IFNAR2 subunits, constitutively associated with the tyrosine kinases TYK2 and JAK1 on its intracellular domain.
Upon interferon binding, these kinases are activated through transphosphorylation, initiating a signal transduction cascade that has become one of the most therapeutically important pathways in autoimmune dermatology: the JAK-STAT pathway. The Janus kinase-signal transducer and activator of transcription pathway translates the extracellular interferon signal into intranuclear gene expression changes that reprogram the transcriptional output of target cells toward a pro-inflammatory, anti-viral phenotype — an "interferon signature" that is now recognized as one of the most consistent biomarker findings in cutaneous and systemic lupus.
Upon interferon binding, these kinases are activated through transphosphorylation, initiating a signal transduction cascade that has become one of the most therapeutically important pathways in autoimmune dermatology: the JAK-STAT pathway. The Janus kinase-signal transducer and activator of transcription pathway translates the extracellular interferon signal into intranuclear gene expression changes that reprogram the transcriptional output of target cells toward a pro-inflammatory, anti-viral phenotype — an "interferon signature" that is now recognized as one of the most consistent biomarker findings in cutaneous and systemic lupus.
The JAK-STAT Pathway: The Molecular Engine of Chronic Inflammation
The JAK-STAT pathway in the context of cutaneous lupus is not a single event but a sustained, self-amplifying process. Interferon-stimulated STAT1 and STAT2 — the principal signal transducers activated by Type I interferon-IFNAR engagement — form heterodimeric and homotrimeric complexes that translocate to the nucleus and drive the transcription of hundreds of interferon-stimulated genes. These genes encode proteins that collectively sustain and amplify the inflammatory state: additional interferons and cytokines, chemokines that recruit further immune effectors to the inflammatory site, molecules that upregulate antigen presentation and lower the threshold for adaptive immune activation, and factors that inhibit the apoptotic clearance of damaged cells — the very clearance defect that initiated the cascade in the first place.
The consequence of sustained JAK-STAT pathway activation in the skin is the chronic, interface-pattern inflammation that characterizes all subtypes of cutaneous lupus histopathologically. Vacuolar degeneration of the basal keratinocyte layer, dermal mucin deposition, a lymphocytic infiltrate hugging the dermal-epidermal junction, and the apoptotic destruction of keratinocytes — collectively termed interface dermatitis — are the microscopic signature of this interferon-driven process. The dermal-epidermal junction is the anatomical focus of inflammation because it is precisely the zone where plasmacytoid dendritic cells aggregate, where interferon concentrations are highest, and where IFNAR-expressing keratinocytes are most directly exposed to the interferon signal.
The therapeutic implications of JAK-STAT pathway centrality in cutaneous lupus pathogenesis are now being actively exploited. JAK inhibitors — small molecules that competitively inhibit the kinase activity of JAK1, JAK2, and TYK2 — interrupt the interferon signaling cascade at the intracellular level, downstream of IFNAR, and have demonstrated meaningful clinical efficacy in several subtypes of cutaneous lupus in early clinical trials. The regulatory approval of deucravacitinib, a selective TYK2 inhibitor, for plaque psoriasis — a condition that shares the JAK-STAT-driven keratinocyte inflammation of cutaneous lupus — has catalyzed interest in TYK2 as a specific therapeutic target in lupus dermatology.
The consequence of sustained JAK-STAT pathway activation in the skin is the chronic, interface-pattern inflammation that characterizes all subtypes of cutaneous lupus histopathologically. Vacuolar degeneration of the basal keratinocyte layer, dermal mucin deposition, a lymphocytic infiltrate hugging the dermal-epidermal junction, and the apoptotic destruction of keratinocytes — collectively termed interface dermatitis — are the microscopic signature of this interferon-driven process. The dermal-epidermal junction is the anatomical focus of inflammation because it is precisely the zone where plasmacytoid dendritic cells aggregate, where interferon concentrations are highest, and where IFNAR-expressing keratinocytes are most directly exposed to the interferon signal.
The therapeutic implications of JAK-STAT pathway centrality in cutaneous lupus pathogenesis are now being actively exploited. JAK inhibitors — small molecules that competitively inhibit the kinase activity of JAK1, JAK2, and TYK2 — interrupt the interferon signaling cascade at the intracellular level, downstream of IFNAR, and have demonstrated meaningful clinical efficacy in several subtypes of cutaneous lupus in early clinical trials. The regulatory approval of deucravacitinib, a selective TYK2 inhibitor, for plaque psoriasis — a condition that shares the JAK-STAT-driven keratinocyte inflammation of cutaneous lupus — has catalyzed interest in TYK2 as a specific therapeutic target in lupus dermatology.
From Molecules to Morphology: The Three Clinical Subtypes
The shared pathogenetic mechanism — UV-triggered nucleic acid release, plasmacytoid dendritic cell activation, interferon production, JAK-STAT signaling, and interface dermatitis — does not produce a single, uniform clinical appearance. The morphology of cutaneous lupus varies with the depth of the inflammatory infiltrate within the skin, the degree of epidermal involvement, the magnitude and chronicity of the immune response, the specific autoantibody profile of the individual patient, and genetic factors that are incompletely understood. These variables generate a clinical spectrum that is conventionally organized into three major morphological categories: acute, subacute, and chronic cutaneous lupus.
Acute Cutaneous Lupus Erythematosus: The Butterfly and Beyond
Acute cutaneous lupus erythematosus is the subtype most intimately associated with systemic disease activity. Its most iconic manifestation is the malar rash — the erythematous, sometimes edematous flush that spreads bilaterally across the cheeks and the bridge of the nose in the configuration that has given rise to the butterfly rash designation. This distribution is not arbitrary: it reflects both the photosensitive nature of the eruption — the malar prominences and nasal dorsum being among the most consistently sun-exposed surfaces of the face — and the tendency of the acute inflammatory process to follow the vascular blush pattern of the central face.
The malar rash of acute cutaneous lupus erythematosus is characteristically erythematous, flat or slightly elevated, warm to the touch, and notably spares the nasolabial folds — a feature that clinically distinguishes it from the erythema of rosacea, which preferentially involves the nasolabial folds, and from seborrheic dermatitis, which concentrates within them. The rash tends to appear acutely, often following sun exposure, and resolves without permanent scarring — a critical distinguishing feature from discoid lupus, which scars by definition.
Acute cutaneous lupus erythematosus extends beyond the malar rash in its more generalized form, producing widespread erythematous macules and papules across the sun-exposed surfaces of the arms, chest, and back in a distribution that can superficially resemble a viral exanthem or drug eruption. Its appearance in a patient with other systemic lupus features — arthritis, serositis, renal disease, hematological abnormalities — is frequently a marker of systemic disease flare, and its treatment is often guided by the management of the underlying systemic disease rather than by topical or local interventions alone.
The malar rash of acute cutaneous lupus erythematosus is characteristically erythematous, flat or slightly elevated, warm to the touch, and notably spares the nasolabial folds — a feature that clinically distinguishes it from the erythema of rosacea, which preferentially involves the nasolabial folds, and from seborrheic dermatitis, which concentrates within them. The rash tends to appear acutely, often following sun exposure, and resolves without permanent scarring — a critical distinguishing feature from discoid lupus, which scars by definition.
Acute cutaneous lupus erythematosus extends beyond the malar rash in its more generalized form, producing widespread erythematous macules and papules across the sun-exposed surfaces of the arms, chest, and back in a distribution that can superficially resemble a viral exanthem or drug eruption. Its appearance in a patient with other systemic lupus features — arthritis, serositis, renal disease, hematological abnormalities — is frequently a marker of systemic disease flare, and its treatment is often guided by the management of the underlying systemic disease rather than by topical or local interventions alone.
Subacute Cutaneous Lupus Erythematosus: The Ring That Wanders
Subacute cutaneous lupus erythematosus occupies an intermediate position in the clinical and serological spectrum of the disease. It presents as recurring, photosensitive, non-scarring eruptions that appear predominantly on the trunk, shoulders, and upper extremities — sun-exposed areas, but characteristically sparing the central face and scalp. The morphology is either annular — ring-shaped plaques with erythematous active borders and central clearing or faint post-inflammatory discoloration — or papulosquamous, resembling psoriasis or eczema with scaly, erythematous patches.
The serological signature of subacute cutaneous lupus erythematosus is its strong association with anti-Ro/SSA antibodies, present in approximately eighty percent of affected patients. This association is not merely a laboratory curiosity — it has pathogenetic significance. Anti-Ro/SSA antibody titers correlate with disease activity, and the Ro antigen — a ribonucleoprotein involved in RNA quality control — is translocated to the keratinocyte surface under conditions of UV stress, where it becomes accessible to the circulating autoantibodies and provides a direct mechanism for autoantibody-mediated keratinocyte injury independent of the plasmacytoid dendritic cell pathway.
A clinically important feature of subacute cutaneous lupus erythematosus is its frequent association with drug exposure. A growing list of medications — including hydrochlorothiazide, calcium channel blockers, angiotensin-converting enzyme inhibitors, proton pump inhibitors, and several chemotherapeutic agents — can induce an SCLE-like eruption in susceptible individuals, presumably by amplifying the photosensitivity or the anti-Ro autoantibody response. Drug-induced SCLE is generally reversible upon discontinuation of the offending agent, and its recognition can spare patients from immunosuppressive therapy directed at a presumed idiopathic process.
The serological signature of subacute cutaneous lupus erythematosus is its strong association with anti-Ro/SSA antibodies, present in approximately eighty percent of affected patients. This association is not merely a laboratory curiosity — it has pathogenetic significance. Anti-Ro/SSA antibody titers correlate with disease activity, and the Ro antigen — a ribonucleoprotein involved in RNA quality control — is translocated to the keratinocyte surface under conditions of UV stress, where it becomes accessible to the circulating autoantibodies and provides a direct mechanism for autoantibody-mediated keratinocyte injury independent of the plasmacytoid dendritic cell pathway.
A clinically important feature of subacute cutaneous lupus erythematosus is its frequent association with drug exposure. A growing list of medications — including hydrochlorothiazide, calcium channel blockers, angiotensin-converting enzyme inhibitors, proton pump inhibitors, and several chemotherapeutic agents — can induce an SCLE-like eruption in susceptible individuals, presumably by amplifying the photosensitivity or the anti-Ro autoantibody response. Drug-induced SCLE is generally reversible upon discontinuation of the offending agent, and its recognition can spare patients from immunosuppressive therapy directed at a presumed idiopathic process.
Discoid Lupus Erythematosus: Chronic, Destructive, and Underestimated
Discoid lupus erythematosus is the chronic, scarring subtype of cutaneous lupus, and it carries the most significant long-term morbidity of the three major forms. Its lesions begin as erythematous papules or plaques with adherent scale, the undersurface of which — when removed — reveals the characteristic carpet-tack appearance caused by follicular plugging. Over weeks to months, the active erythematous border slowly expands while the central zone evolves through atrophy, dyspigmentation, and ultimately permanent scarring — a progression that, unlike the reversible changes of acute or subacute cutaneous lupus, cannot be undone by any currently available therapy.
The face, scalp, and external ears are the most commonly affected sites, with the scalp involvement carrying particularly devastating consequences. Discoid lupus of the scalp destroys hair follicles within the zone of inflammation, replacing the follicular architecture with fibrotic scar tissue and producing permanent, non-reversible scarring alopecia. Unlike the diffuse, telogen effluvium-type hair loss of systemic lupus — which is reversible with disease control — the follicular destruction of discoid lupus is irreversible, and the cosmetic and psychological impact for affected patients, particularly women of reproductive age who represent the demographic most commonly affected, can be profound and lasting.
Histopathologically, discoid lupus erythematosus demonstrates the interface dermatitis pattern of all cutaneous lupus subtypes, but with additional features reflecting the chronicity and depth of the inflammatory process: a dense, lichenoid lymphocytic infiltrate extending deep into the dermis and around dermal appendages, basement membrane thickening with PAS-positive deposits, prominent dermal mucin deposition, and the follicular plugging and hyperkeratosis that reflect the keratinocyte response to sustained inflammatory injury. The direct immunofluorescence study — the lupus band test — reveals granular deposits of immunoglobulin and complement at the dermal-epidermal junction in both lesional and, in systemic lupus, non-lesional skin, providing a histological window into the autoantibody-mediated component of the pathogenetic cascade.
The relationship between discoid lupus erythematosus and systemic disease is a question of clinical importance that remains only partially resolved. Approximately five percent of patients with isolated discoid lupus erythematosus will develop systemic lupus erythematosus over time, a risk that is higher in patients with disseminated discoid lesions involving sun-protected as well as sun-exposed sites, in those with positive antinuclear antibodies or anti-dsDNA antibodies, and in those with hematological or renal abnormalities on laboratory evaluation. Conversely, roughly twenty percent of patients with systemic lupus erythematosus will have discoid lesions at some point in their disease course. Regular systemic monitoring — urinalysis, complete blood count, complement levels, anti-dsDNA titers — is therefore warranted in all patients with established discoid lupus erythematosus, regardless of their current systemic symptoms.
The face, scalp, and external ears are the most commonly affected sites, with the scalp involvement carrying particularly devastating consequences. Discoid lupus of the scalp destroys hair follicles within the zone of inflammation, replacing the follicular architecture with fibrotic scar tissue and producing permanent, non-reversible scarring alopecia. Unlike the diffuse, telogen effluvium-type hair loss of systemic lupus — which is reversible with disease control — the follicular destruction of discoid lupus is irreversible, and the cosmetic and psychological impact for affected patients, particularly women of reproductive age who represent the demographic most commonly affected, can be profound and lasting.
Histopathologically, discoid lupus erythematosus demonstrates the interface dermatitis pattern of all cutaneous lupus subtypes, but with additional features reflecting the chronicity and depth of the inflammatory process: a dense, lichenoid lymphocytic infiltrate extending deep into the dermis and around dermal appendages, basement membrane thickening with PAS-positive deposits, prominent dermal mucin deposition, and the follicular plugging and hyperkeratosis that reflect the keratinocyte response to sustained inflammatory injury. The direct immunofluorescence study — the lupus band test — reveals granular deposits of immunoglobulin and complement at the dermal-epidermal junction in both lesional and, in systemic lupus, non-lesional skin, providing a histological window into the autoantibody-mediated component of the pathogenetic cascade.
The relationship between discoid lupus erythematosus and systemic disease is a question of clinical importance that remains only partially resolved. Approximately five percent of patients with isolated discoid lupus erythematosus will develop systemic lupus erythematosus over time, a risk that is higher in patients with disseminated discoid lesions involving sun-protected as well as sun-exposed sites, in those with positive antinuclear antibodies or anti-dsDNA antibodies, and in those with hematological or renal abnormalities on laboratory evaluation. Conversely, roughly twenty percent of patients with systemic lupus erythematosus will have discoid lesions at some point in their disease course. Regular systemic monitoring — urinalysis, complete blood count, complement levels, anti-dsDNA titers — is therefore warranted in all patients with established discoid lupus erythematosus, regardless of their current systemic symptoms.
Diagnosis: Integrating the Clinical and the Molecular
The diagnosis of cutaneous lupus erythematosus is fundamentally clinical, supported by histopathological confirmation and serological evaluation. No single laboratory test or biopsy finding is pathognomonic in isolation; the diagnosis emerges from the integration of morphological pattern, anatomical distribution, histological findings, immunofluorescence results, and serological profile.
The clinical encounter should establish the photosensitive character of the eruption, its distribution relative to sun-exposed sites, the presence or absence of scarring, and any history of prior similar episodes. A detailed medication review is essential in all patients with newly diagnosed subacute cutaneous lupus erythematosus. Skin biopsy should be obtained from the active border of a well-developed lesion, submitted for routine histopathology in formalin and for direct immunofluorescence in Michel's transport medium. Serological evaluation should include antinuclear antibody by immunofluorescence, followed by specific autoantibody testing for anti-dsDNA, anti-Sm, anti-Ro/SSA, and anti-La/SSB, with anti-Ro/SSA being the most clinically relevant for subacute cutaneous lupus.
The differential diagnosis of cutaneous lupus erythematosus is broad and reflects the disease's status as an imitator. The butterfly rash of acute cutaneous lupus is most commonly confused with rosacea, seborrheic dermatitis, and photodermatitis. Subacute cutaneous lupus erythematosus mimics psoriasis, tinea corporis, and nummular eczema. Discoid lupus erythematosus must be distinguished from lichen planus, lichen planopilaris, pseudopelade of Brocq, and, in its hypertrophic variant, squamous cell carcinoma — a diagnostic exclusion that has particular urgency given that longstanding discoid lupus erythematosus carries a small but established risk of malignant transformation within chronic scar tissue.
The clinical encounter should establish the photosensitive character of the eruption, its distribution relative to sun-exposed sites, the presence or absence of scarring, and any history of prior similar episodes. A detailed medication review is essential in all patients with newly diagnosed subacute cutaneous lupus erythematosus. Skin biopsy should be obtained from the active border of a well-developed lesion, submitted for routine histopathology in formalin and for direct immunofluorescence in Michel's transport medium. Serological evaluation should include antinuclear antibody by immunofluorescence, followed by specific autoantibody testing for anti-dsDNA, anti-Sm, anti-Ro/SSA, and anti-La/SSB, with anti-Ro/SSA being the most clinically relevant for subacute cutaneous lupus.
The differential diagnosis of cutaneous lupus erythematosus is broad and reflects the disease's status as an imitator. The butterfly rash of acute cutaneous lupus is most commonly confused with rosacea, seborrheic dermatitis, and photodermatitis. Subacute cutaneous lupus erythematosus mimics psoriasis, tinea corporis, and nummular eczema. Discoid lupus erythematosus must be distinguished from lichen planus, lichen planopilaris, pseudopelade of Brocq, and, in its hypertrophic variant, squamous cell carcinoma — a diagnostic exclusion that has particular urgency given that longstanding discoid lupus erythematosus carries a small but established risk of malignant transformation within chronic scar tissue.
Treatment: Blocking the Cascade at Multiple Points
The treatment of cutaneous lupus erythematosus is organized around two fundamental principles: prevention of the UV trigger and suppression of the autoimmune inflammatory cascade. Photoprotection — broad-spectrum sunscreen with SPF 50 or higher applied daily to all exposed sites, protective clothing, avoidance of peak UV hours, and the use of UV-blocking window films — is not optional adjunctive advice but a primary therapeutic intervention. The evidence that consistent photoprotection reduces disease flare frequency, lesion area, and systemic disease activity in photosensitive lupus patients is robust, and its failure to be implemented is one of the most common drivers of inadequately controlled cutaneous lupus in clinical practice.
Pharmacological management begins with topical and intralesional corticosteroids for localized disease and topical calcineurin inhibitors — tacrolimus and pimecrolimus — as steroid-sparing alternatives particularly suited to the sensitive skin of the face and skin folds. The antimalarial hydroxychloroquine remains the systemic agent of first choice for all subtypes of cutaneous lupus, with a safety profile accumulated over decades of use and evidence supporting both its anti-inflammatory and its photoprotective mechanisms of action at the cellular level.
For hydroxychloroquine-refractory disease, the therapeutic armamentarium includes systemic corticosteroids for acute control, methotrexate, mycophenolate mofetil, dapsone, and retinoids. Belimumab — a monoclonal antibody targeting B-lymphocyte stimulator — is approved for systemic lupus erythematosus and has demonstrated cutaneous benefit in patients with active skin disease. Anifrolumab, an anti-IFNAR1 monoclonal antibody that directly blocks the interferon receptor, represents a mechanistically elegant targeted intervention that acts precisely at the signaling node illustrated in Figure 1, and it has demonstrated significant clinical benefit in cutaneous lupus in the phase III TULIP trials, including meaningful improvements in the Cutaneous Lupus Erythematosus Disease Area and Severity Index scores.
The emerging JAK inhibitor class — particularly TYK2 inhibitors that target the kinase immediately downstream of IFNAR — offers the prospect of oral, once-daily agents that interrupt the JAK-STAT cascade with a level of molecular specificity that conventional immunosuppressants cannot achieve. Clinical trials of deucravacitinib in systemic lupus erythematosus are ongoing, and their cutaneous endpoints will be closely watched by the lupus dermatology community.
Pharmacological management begins with topical and intralesional corticosteroids for localized disease and topical calcineurin inhibitors — tacrolimus and pimecrolimus — as steroid-sparing alternatives particularly suited to the sensitive skin of the face and skin folds. The antimalarial hydroxychloroquine remains the systemic agent of first choice for all subtypes of cutaneous lupus, with a safety profile accumulated over decades of use and evidence supporting both its anti-inflammatory and its photoprotective mechanisms of action at the cellular level.
For hydroxychloroquine-refractory disease, the therapeutic armamentarium includes systemic corticosteroids for acute control, methotrexate, mycophenolate mofetil, dapsone, and retinoids. Belimumab — a monoclonal antibody targeting B-lymphocyte stimulator — is approved for systemic lupus erythematosus and has demonstrated cutaneous benefit in patients with active skin disease. Anifrolumab, an anti-IFNAR1 monoclonal antibody that directly blocks the interferon receptor, represents a mechanistically elegant targeted intervention that acts precisely at the signaling node illustrated in Figure 1, and it has demonstrated significant clinical benefit in cutaneous lupus in the phase III TULIP trials, including meaningful improvements in the Cutaneous Lupus Erythematosus Disease Area and Severity Index scores.
The emerging JAK inhibitor class — particularly TYK2 inhibitors that target the kinase immediately downstream of IFNAR — offers the prospect of oral, once-daily agents that interrupt the JAK-STAT cascade with a level of molecular specificity that conventional immunosuppressants cannot achieve. Clinical trials of deucravacitinib in systemic lupus erythematosus are ongoing, and their cutaneous endpoints will be closely watched by the lupus dermatology community.
Conclusion: A Disease Illuminated by the Mechanism That Drives It
Cutaneous lupus erythematosus is a disease of molecular mistaken identity — an immune system that has lost the ability to distinguish its own nucleic acids from those of a pathogen, and that responds to that confusion with chronic, destructive inflammation targeting the very tissue through which the error was first made visible. The pathogenetic cascade, from UV photodamage through nucleic acid release and plasmacytoid dendritic cell activation to interferon amplification and JAK-STAT-driven interface dermatitis, is one of the best-characterized autoimmune mechanisms in all of dermatology.
The clinical heterogeneity of the disease — the transient flush of acute cutaneous lupus, the wandering annular plaques of subacute cutaneous lupus, the progressive scarring of discoid lupus — is not a contradiction of the shared mechanism but a consequence of the many variables that modulate its expression in individual patients: the depth of dermal infiltration, the chronicity of the inflammatory stimulus, the specific autoantibody profile, and the genetic architecture of immune regulation that each patient brings to the encounter.
For the clinician, this mechanistic clarity carries both diagnostic and therapeutic implications. Diagnostically, it explains why photosensitivity is not merely a feature of cutaneous lupus but its fundamental pathological initiator — and why photoprotection is not supportive care but disease modification. Therapeutically, it reveals a cascade with multiple intervention points, from the UV exposure that starts it to the interferon receptor that amplifies it to the JAK kinases that propagate it — a cascade that the newest generation of targeted biologics and small molecules is beginning to interrupt with a precision that was not possible when patients with this disease were first taught to stay out of the sun.
The clinical heterogeneity of the disease — the transient flush of acute cutaneous lupus, the wandering annular plaques of subacute cutaneous lupus, the progressive scarring of discoid lupus — is not a contradiction of the shared mechanism but a consequence of the many variables that modulate its expression in individual patients: the depth of dermal infiltration, the chronicity of the inflammatory stimulus, the specific autoantibody profile, and the genetic architecture of immune regulation that each patient brings to the encounter.
For the clinician, this mechanistic clarity carries both diagnostic and therapeutic implications. Diagnostically, it explains why photosensitivity is not merely a feature of cutaneous lupus but its fundamental pathological initiator — and why photoprotection is not supportive care but disease modification. Therapeutically, it reveals a cascade with multiple intervention points, from the UV exposure that starts it to the interferon receptor that amplifies it to the JAK kinases that propagate it — a cascade that the newest generation of targeted biologics and small molecules is beginning to interrupt with a precision that was not possible when patients with this disease were first taught to stay out of the sun.
Urinary Tract Infection Updates: Current Clinical and Public Health Developments
Urinary tract infection remains one of the most common bacterial infections globally, with current updates focusing on antimicrobial resistance, diagnostic stewardship, recurrent infection management, catheter-associated prevention, and emerging non-antibiotic strategies. This article reviews recent developments relevant to clinicians, researchers, and public health professionals.
Top 5 Vegetables That May Enhance Immune Function: An Evidence-Based Nutritional Overview
Dietary patterns rich in vegetables are consistently associated with improved health outcomes, including support of normal immune function. This article reviews five vegetables—broccoli, spinach, garlic, carrots, and red bell peppers—that may contribute to immune resilience through their content of vitamins, minerals, antioxidants, and bioactive phytochemicals.


Understanding Stunted Children: Chronic Growth Failure, Early-Life Risks, and Why It Matters Beyond Height
Stunting in children refers to impaired linear growth resulting from chronic undernutrition, repeated infection, and unfavorable early-life conditions. More than a matter of short stature, stunting reflects a broader process of biological and developmental disadvantage that can affect cognitive outcomes, school performance, and long-term health.
Blood Pressure Monitoring as a Public Health Priority: Strengthening Early Detection and Long-Term Cardiovascular Prevention
Blood pressure monitoring remains one of the most practical and impactful tools in public health for identifying hypertension early, guiding treatment decisions, and reducing long-term cardiovascular risk. Wider adoption of accurate office, community, and home-based monitoring strategies could significantly improve prevention of stroke, heart disease, kidney damage, and premature mortality.

